Rice data
Download the Rice Diversity Panel data RiceDiversity.44K.MSU6.Genotypes_PLINK.zip from http://ricediversity.org/data/sets/44kgwas/.
Genotypic data
rice <- read.table("http://www.ricediversity.org/data/sets/44kgwas/RiceDiversity_44K_Phenotypes_34traits_PLINK.txt", header=TRUE, stringsAsFactors = FALSE, sep = "\t")
rice <- rice[, 1:3]
set.seed(123579)
for(i in 1:1000){
## add a fake genotype assuming the pop in HWE
p = runif(n=1, min=0.05, max=0.5)
q = 1-p
rice$geno <- sample(c(0,1,2), size=nrow(rice), prob=c(p^2, 2*p*q, q^2), replace=TRUE)
names(rice)[ncol(rice)] <- paste0("m", i)
}Check the genotype, i.e., the minor allele frequency
##
## 0 1 2
## 4 61 348## [1] 0.08353511Apply the method to all the markers:
# A function to calculate minor allele frequencies for all the markers
maf <- function(rice){
df <- data.frame()
for(i in 4:ncol(rice)){
tb <- as.data.frame(table(rice[,i]))
a1 <- 2*tb$Freq[1] + tb$Freq[2]
frq <- data.frame(marker=names(rice)[i], maf=a1/(2*nrow(rice)))
if(frq$maf > 0.5){
frq$maf <- 1 - frq$maf
}
df <- rbind(df, frq)
}
return(df)
}
out <- maf(rice)
hist(out$maf, breaks=30, col="blue", xlim=c(0, 0.5), main="MAF distribution", xlab="freq")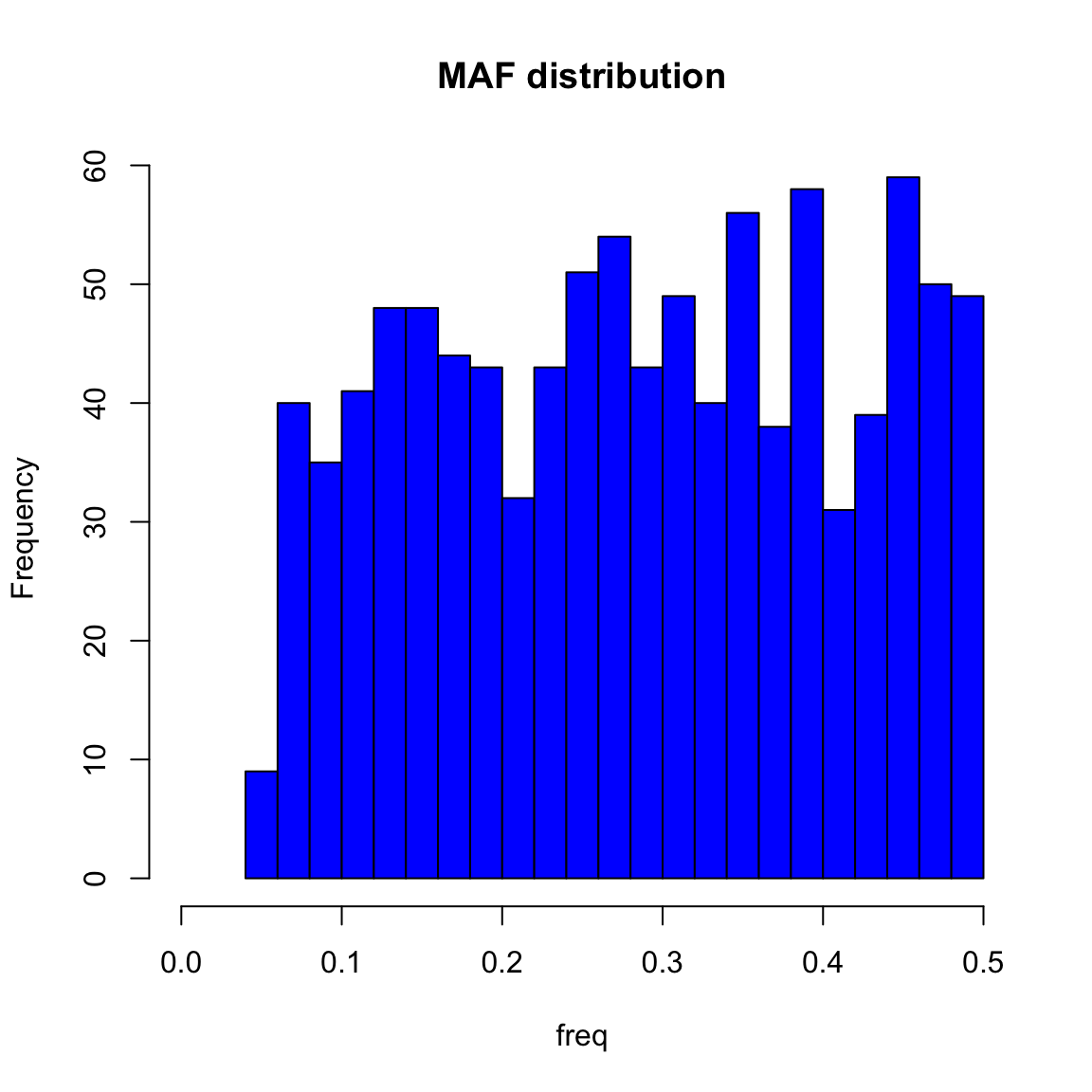
Phenotype data
Simulate QTLs controlling flowering time
Simulate phenotype using genotype data given number of QTLs and heritability.
sim_qtl_pheno <- function(geno, h2, nqtl){
#' @param geno genotype data, col1=genoid, col2 and after=snpid, coding: 0,1,2 (no missing data allowed) [data.frame].
#' @param h2 Broad sense heritability of the trait [numeric(0-1)].
#' @param nqtl number of QTL [interger].
#' @param distribution [character=norm]
#'
#' @return return A list of many values.
#'
#' @examples
#' geno <- read.table("data/geno.txt", header=TRUE)
#' pheno <- sim_qtl_pheno(geno, h2=0.7, alpha=0.5, nqtl=10, distribution="norm")
#' y <- pheno[['y']]
X <- geno[, -1]
n <- nrow(X)
m <- ncol(X)
set.seed(1237)
#Sampling QTN
QTN.position <- sample(m, nqtl, replace=F)
SNPQ <- as.matrix(X[, QTN.position])
message(sprintf("### [simpheno], read in [ %s ] SNPs for [ %s ] plants and simulated [ %s ] QTLs",
m, n, nqtl))
#Simulate phenotype
addeffect <- rnorm(nqtl,0,1)
effect <- SNPQ%*%addeffect
effectvar <- var(effect)
residualvar <- (effectvar - h2*effectvar)/h2
residual <- rnorm(n, 0, sqrt(residualvar))
y <- effect + residual
return(data.frame(y=y, SNPQ=SNPQ))
}Apply the QTL simulation function to simulate phenotype with the given values of the h2 and number of QTLs nqtl.
## ### [simpheno], read in [ 1000 ] SNPs for [ 413 ] plants and simulated [ 2 ] QTLs# assuming the values deviated from 100 days
ph$y <- ph$y+100
rice$Flowering.time.at.Arkansas <- ph$y
names(rice)[3] <- "ft"Plot the phenotypic distribution:
# phenotypes
hist(rice$ft, breaks=30, col="blue",
main="Distribution of the flowering time values",
xlab="flowering time (days)")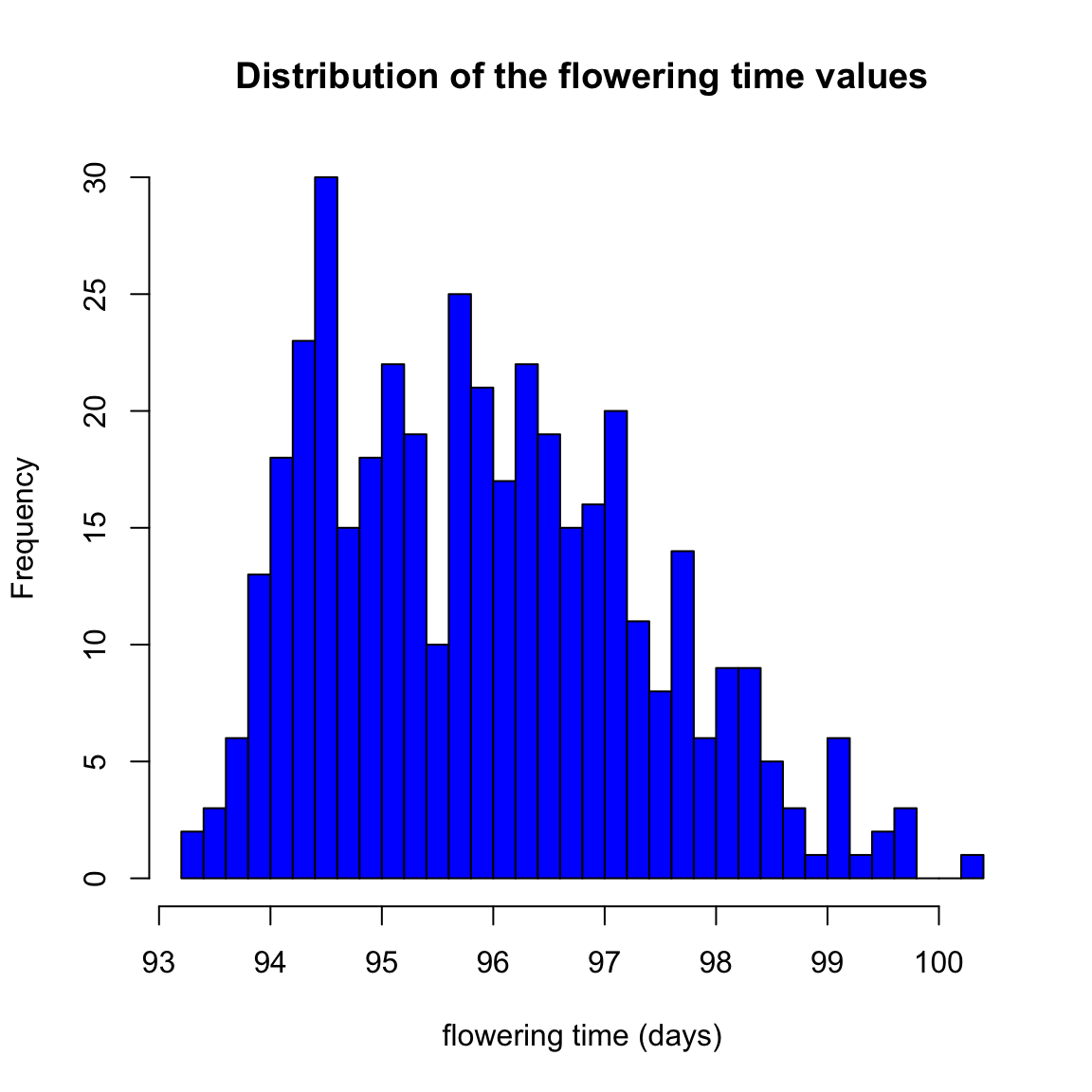
Average effect of the markers
\(Y = G + E\)
\(Y = \sum_{i=1}^n{G_i} + E\) for a population with \(i\) markers
\(G = A + D\)
If we find \(A_i\) for \(i\)th marker, then
\(\hat{Y} = \sum_{i=1}^n{A_i}\)
Single locus
gfunction <- function(a=1, d=0, p=1/2){
# a: additive effect
# d: dominant effect
# p: allele frequency for the A1 allele
q = 1-p
# allele sub effect
alpha <- a + d*(q-p)
a1a1 <- 2*alpha*q
a1a2 <- (q-p)*alpha
a2a2 <- -2*p*alpha
# population mean
M <- a*(p-q) + 2*d*p*q
# return a data.frame with genotype values and breeding values
return(data.frame(N1=c(0,1,2), gv=c(-a-M,d-M,a-M), bv=c(a2a2, a1a2, a1a1)))
}- A1A1 -> 2
- A1A2 -> 1
- A2A2 -> 0
A1A1 <- mean(subset(rice, m1 == 2)$ft, na.rm=TRUE)
A1A2 <- mean(subset(rice, m1 == 1)$ft, na.rm=TRUE)
A2A2 <- mean(subset(rice, m1 == 0)$ft, na.rm=TRUE)
### mid point
m <- (A1A1 + A2A2)/2
a <- A1A1 - m
d <- A1A2 - m
df <- as.data.frame(table(rice$m1))
# allele freq for A1 (2 coding)
p <- (2*df[df$Var1==2,]$Freq + df[df$Var1==1,]$Freq)/(2*sum(df$Freq))
out <- gfunction(a=a, d=d, p=p)
avg <- (out$bv[1] - out$bv[3])/2Next we wrap it into a function to calcuate the avg effect for all the markers
avgfun <- function(rice){
outdf <- data.frame()
for(i in 4:ncol(rice)){
A1A1 <- mean(subset(rice, rice[,i] == 2)$ft, na.rm=TRUE)
A1A2 <- mean(subset(rice, rice[,i] == 1)$ft, na.rm=TRUE)
A2A2 <- mean(subset(rice, rice[,i] == 0)$ft, na.rm=TRUE)
### mid point
m <- (A1A1 + A2A2)/2
a <- A1A1 - m
d <- A1A2 - m
df <- as.data.frame(table(rice[,i]))
# allele freq for A1 (2 coding)
p <- (2*df[df$Var1==2,]$Freq + df[df$Var1==1,]$Freq)/(2*sum(df$Freq))
out <- gfunction(a=a, d=d, p=p)
avg <- (out$bv[1] - out$bv[3])/2
tem <- data.frame(snp=names(rice)[i], eff=avg)
outdf <- rbind(outdf, tem)
}
return(outdf)
}
outdf <- avgfun(rice)
plot(1:1000, abs(outdf$eff))Additive and dominance variance
Breeder’s equation
\[\begin{align*} R & = ih^2\sigma_P/L \\ & ih\sigma_A/L \\ \end{align*}\]
Because \(G = A + D\), then
\[\begin{align*} \sigma_G^2 & = \sigma_A^2 + \sigma_D^2 + 2\sigma_{A, D} \end{align*}\]
And \(\sigma_{A, D}=0\) in a HWE population, therefore,
\[\begin{align*} \sigma_G^2 & = \sigma_A^2 + \sigma_D^2 \end{align*}\]
| Genotype | Freq | Breeding Value | \(A^2\) | Dominance Deviation | \(D^2\) | |
|---|---|---|---|---|---|---|
| \(A_1A_1\) | \(p^2\) | \(2q\alpha\) | \((2q\alpha)^2\) | \(-2q^2d\) | \((-2q^2d)^2\) | |
| \(A_1A_2\) | \(2pq\) | \((q-p)\alpha\) | \((q-p)^2\alpha^2\) | \(2pqd\) | \((2pqd)^2\) | |
| \(A_2A_2\) | \(q^2\) | \(-2p\alpha\) | \((-2p\alpha)^2\) | \(-2p^2d\) | \((-2p^2d)^2\) |
The additive and dominance genetic variance in a HWE population is:
\[\begin{align*} \sigma_A^2 & = 2pq(a + d(q-p))^2 \\ \sigma_D^2 & = (2pqd)^2 \\ \end{align*}\]
Write the Vg function
\[\begin{align*} \sigma_A^2 & = 2pq(a + d(q-p))^2 \\ \sigma_D^2 & = (2pqd)^2 \\ \end{align*}\]
vfun <- function(a=1, dod=0, p=seq(0,1, by=0.01)){
# a: additive value, [num, =1]
# dod: degree of dominance, [num, =0]
# p: allele frequency of the A1 allele, [vector, =seq(0,1, by=0.01)]
#a = 1
d = dod*a #<< get the dominance value
q = 1- p
df <- data.frame(p=p,
va=2*p*q*(a + d*(q-p))^2,
vd=(2*p*q*d)^2)
df$vg <- df$va + df$vd
return(df)
}Heritability calculation
h2fun <- function(rice){
outdf <- data.frame()
for(i in 4:ncol(rice)){
A1A1 <- mean(subset(rice, rice[,i] == 2)$ft, na.rm=TRUE)
A1A2 <- mean(subset(rice, rice[,i] == 1)$ft, na.rm=TRUE)
A2A2 <- mean(subset(rice, rice[,i] == 0)$ft, na.rm=TRUE)
### mid point
m <- (A1A1 + A2A2)/2
a <- A1A1 - m
d <- A1A2 - m
df <- as.data.frame(table(rice[,i]))
# allele freq for A1 (2 coding)
p <- (2*df[df$Var1==2,]$Freq + df[df$Var1==1,]$Freq)/(2*sum(df$Freq))
out <- vfun(a=a, dod=d/a, p=p)
out$snp <- names(rice)[i]
outdf <- rbind(outdf, out)
}
return(outdf)
}
outdf2 <- h2fun(rice)
plot(1:1000, abs(outdf2$vg), ylab="Variance explained by markers")Broad sense heritability
\(H^2 = V_G / V_P\)
Narrow sense heritability
\(h^2 = V_A / V_P\)
Breeder’s equation
Intensity of selection ( \(i\) )
# function for selection intensity
ifun <- function(p=0.5){
x=qnorm(p=(1-p)) # get the truncation point
z=dnorm(x) # get z
return(z/p) # get i
}
p <- seq(0.0001, 1, by=0.0001)
i <- ifun(p)
plot(p, i)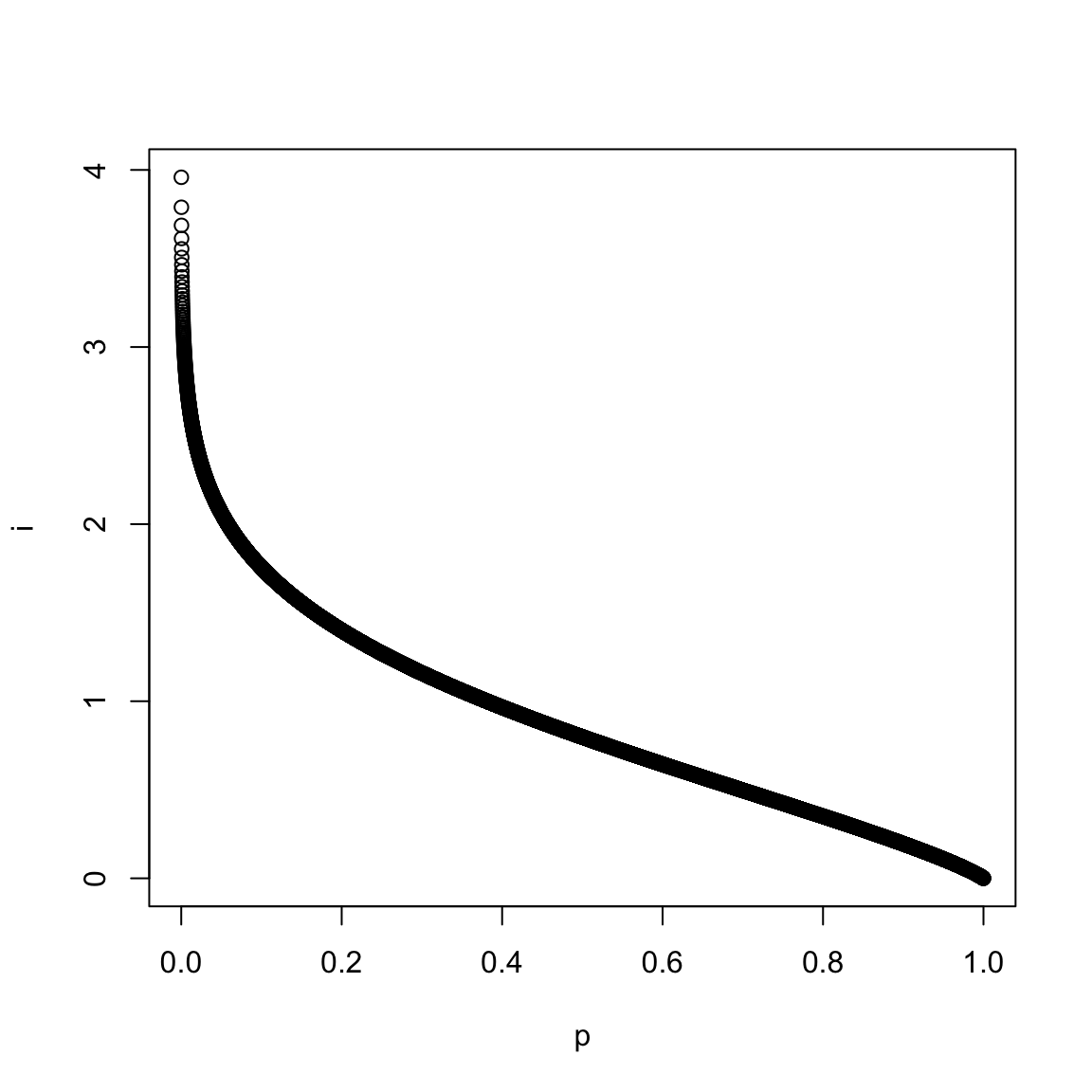
\[\begin{align*} R & = ih^2\sigma_P/L \\ & ih\sigma_A/L \\ \end{align*}\]
# response to selection
Rfun <- function(perc=0.05, h2, va){
i <- ifun(p=perc)
h <- sqrt(h2)
return(i*h*sqrt(va))
}
outR <- Rfun(perc=0.05, h2=0.9, va=2.327923)
50/outR## [1] 16.74656A sib analysis
A swine breeder collected a set of data from a sib design: - 2 sires + 34 dams
- Each pair of parents has four offspring
- Measured body weight of the newborns
- Calculate the estimate of heritability
- Comment on any possible bias in the estimated heritability
Get the ANOVA table
\(Y = S + D + E\)
\(V_P = V_S + V_D + V_E\)
## Df Sum Sq Mean Sq F value Pr(>F)
## sire 1 6331 6331 17.978 3.73e-05 ***
## dam 33 24216 734 2.084 0.00141 **
## Residuals 163 57405 352
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Variance components in a sib design
| Source | df | Sums of Squares | MS | E(MS) |
|---|---|---|---|---|
| Sires | s-1 | \(dn\sum\limits_{i=1}^s(\bar{p}_i - \bar{p})^2\) | \(MS_s\) | \(= \sigma_w^2 + n\sigma_d^2 + dn\sigma_s^2\) |
| Dams (Sires) | s(d-1) | \(n\sum\limits_{i=1}^s\sum\limits_{j=1}^d(\bar{p}_{ij} - \bar{p}_i)^2\) | \(MS_d\) | \(= \sigma_w^2 + n\sigma_d^2\) |
| Sibs (Dams) | sd(n-1) | \(\sum\limits_{i=1}^s\sum\limits_{j=1}^d\sum\limits_{k=1}^n(p_{ijk} - \bar{p}_{ij})^2\) | \(MS_w\) | \(= \sigma_w^2\) |
Summary of the variance components
A key concept in the ANOVA is that the variance between-family (group) is equal to the covariance within-family (group).
| Observational | Covariance and causal components | estimated values |
|---|---|---|
| Sires | \(\sigma_s^2 = Cov(HS)\) | \(=\frac{1}{4}\sigma_A^2\) |
| Dams | \(\sigma_d^2 = Cov(FS) - Cov(HS)\) | \(=\frac{1}{4}\sigma_A^2 + \frac{1}{4}\sigma_D^2\) |
| Progeny | \(\sigma_w^2 = V_P - Cov(FS)\) | \(= \frac{1}{2}\sigma_A^2 +\frac{3}{4}\sigma_D^2\) |
| Total | \(\sigma_T^2 = V_P = \sigma_s^2 + \sigma_d^2 + \sigma_w^2\) | \(=\sigma_A^2 + \sigma_D^2 + \sigma_E^2\) |
| Sires + Dams | \(\sigma_s^2 + \sigma_d^2 = Cov(FS)\) | \(=\frac{1}{2}\sigma_A^2 + \frac{1}{4}\sigma_D^2\) |
\(\sigma_s^2 = Cov(HS) = 1/4 \sigma_A^2\)
\(h^2 = V_A/V_P\)
## [1] 0.3462044Vd
\(\sigma_d^2 = Cov(FS) - Cov(HS) = 1/4 \sigma_A^2 + 1/4 \sigma_D^2\)